Research
| Epigenetic Regulations |
 |
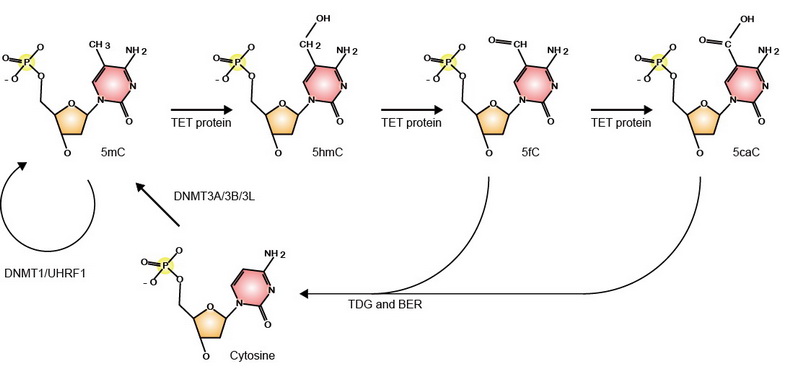
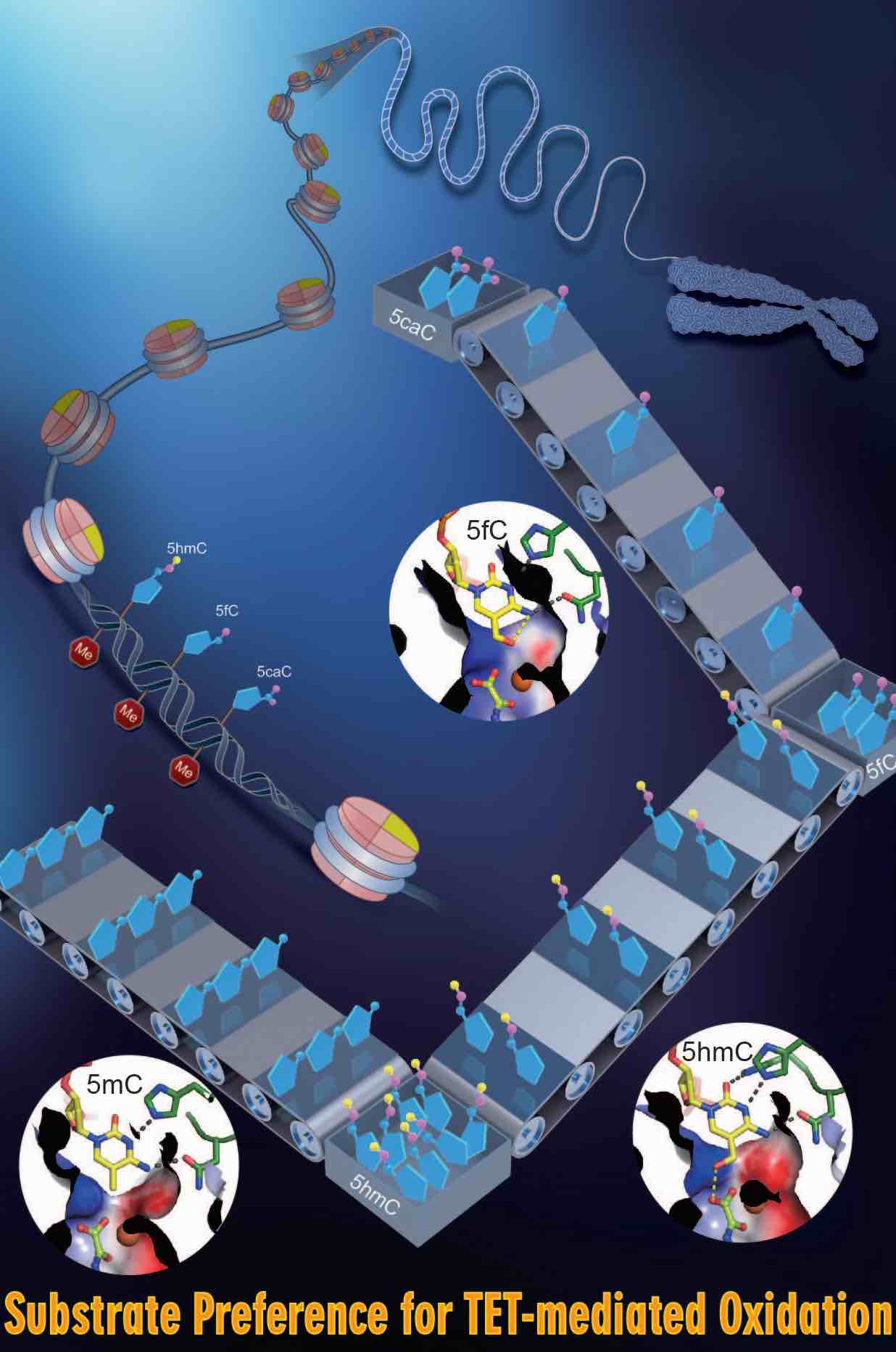
DNA methylation is an important epigenetic modification and essential for various developmental processes through regulating gene expression, genomic imprinting, and epigenetic inheritance. Mammalian genomic DNA methylation is established during embryogenesis by de novo DNA methyltransferases, DNMT3A and DNMT3B. The pattern of DNA methylation is maintained by DNMT1 and UHRF1. Ten-Eleven Translocation (TET) proteins were found to be essential for DNA demethylation. TET1/2/3 oxidize 5mC to 5hmC, 5fC, and 5caC, which are replaced by unmethylated cytosine through active and passive mechanisms.
Our systematic studies reveal the underlying mechanisms for catalysis, substrate recognition, and regulation of enzymatic activity of these epigenetic regulators and provide structural basis for designing of specific inhibitors or activators for functional studies and potential therapeutic applications.
|
Establishment of DNA Methylation
|
| Autoinhibition and H3-Induced Activation of DNMT3A |
|
 MOVIE MOVIE |
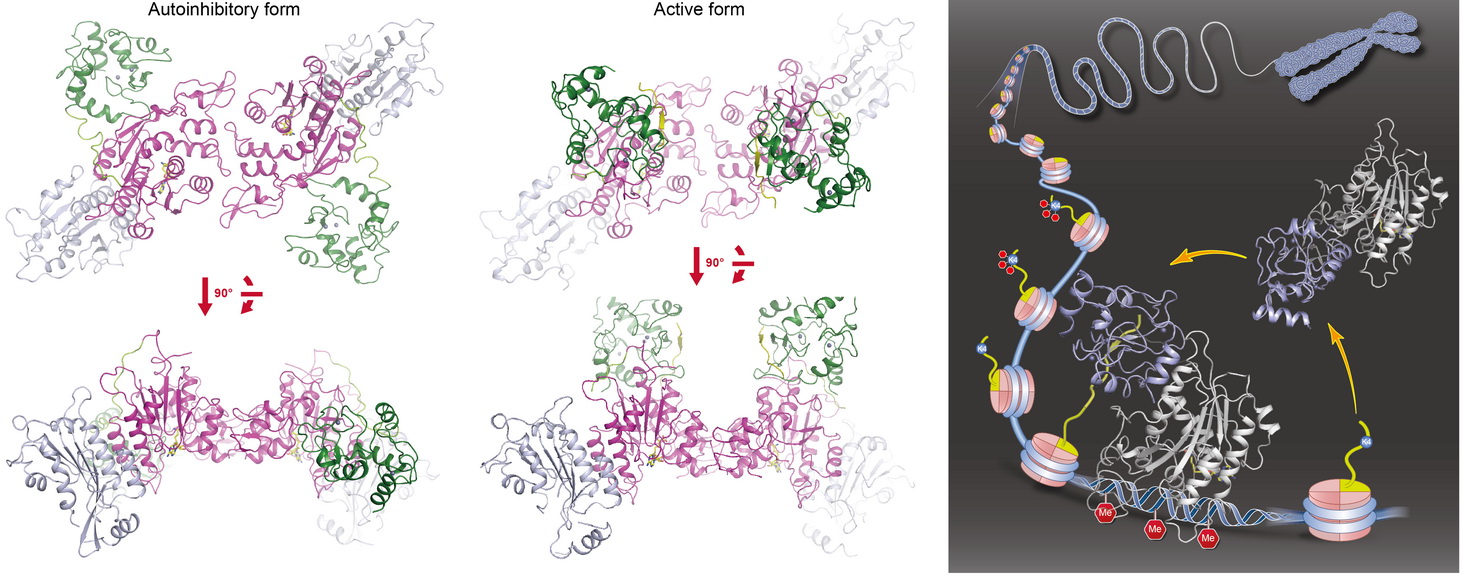
We found that DNMT3A exists in an autoinhibitory form and histone H3 tail stimulates its activity in a DNMT3L-independent manner. We determined the crystal structures of DNMT3A-DNMT3L (autoinhibitory form, left panel) and DNMT3A-DNMT3L-H3 (active form, middle panel) complexes ( Nature, 2015a). The analyses indicate that the ADD domain of DNMT3A interacts with and inhibits enzymatic activity of the catalytic domain (CD) through blocking its DNA-binding affinity. Histone H3 (but not H3K4me3) disrupts ADD-CD interaction, induces a large movement of the ADD domain, and thus releases the autoinhibition of DNMT3A. We propose a working model (right panel) for histone H3-induced dynamic regulation of the de novo DNA methyltransferase. The finding adds another layer of regulation of DNA methylation to ensure that the enzyme is mainly activated at proper targeting loci when unmethylated H3K4 is present, and strongly supports a negative correlation between H3K4me3 and DNA methylation across mammalian genome. Reviewed by:Faculty of 1000 |
Mechanism for DNA Demethylation
|
| Molecular Mechanism for TET-Mediated 5mC Oxidation |
|
MOVIE |
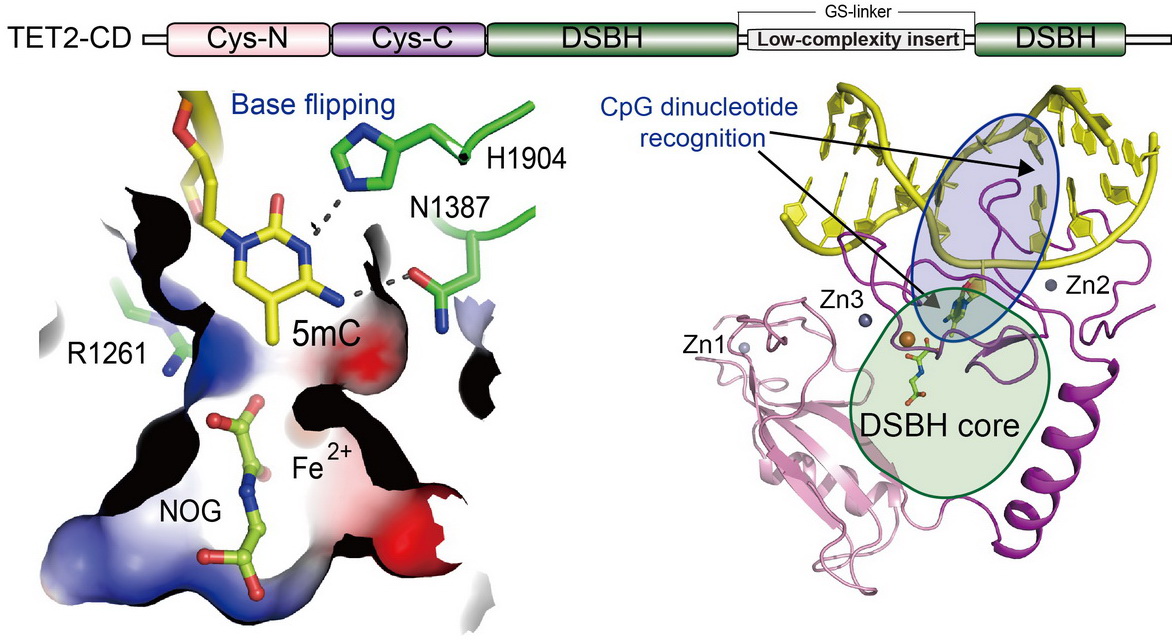
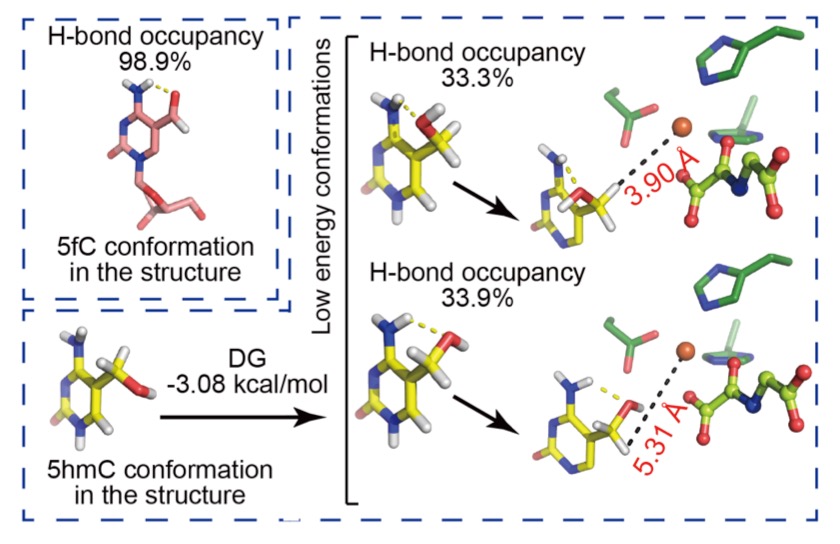
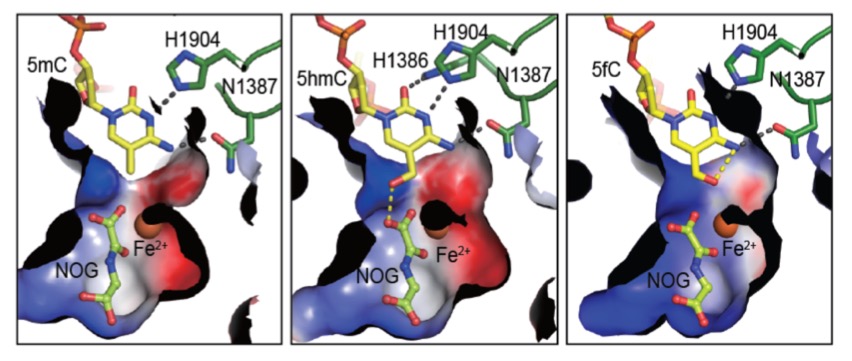
TET proteins oxidize 5-methylcytosine (5mC) on DNA and play important roles in various biological processes. Mutations of TET2 are frequently observed in myeloid malignance. We determined the crystal structure of human TET2 bound to methylated DNA at 2.02 ? resolution ( Cell, 2013). The structure shows that two zinc fingers bring the Cys-rich and DSBH domains together to form a compact catalytic domain. The Cys-rich domain stabilizes the DNA above the DSBH core. TET2 specifically recognizes CpG dinucleotide and shows substrate preference for 5mC in a CpG context. 5mC is inserted into the catalytic cavity with the methyl group orientated to catalytic Fe(II) for reaction. The methyl group is not involved in TET2-DNA contacts so that the catalytic cavity allows TET2 to accommodate 5mC derivatives for further oxidation. Mutations of Fe(II)/NOG-chelating, DNA-interacting and zinc-chelating residues are frequently observed in human cancers. The study provides a structural basis for understanding the mechanisms of TET-mediated 5mC oxidation. Reviewed by:Faculty of 1000, Nature China, Preview in Cell
|
Maintenance of DNA Methylation |
| Mechanism for USP7-mediated DNMT1 stabilization by acetylation |
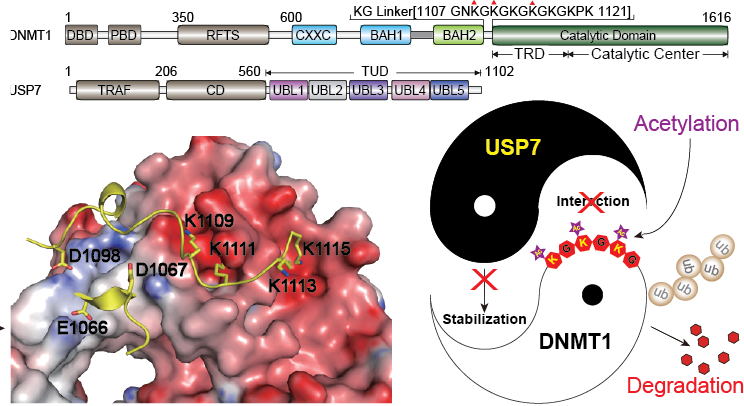
DNMT1 is an important epigenetic regulator that plays a key role in the maintenance of DNA methylation. We determined the crystal structure of DNMT1 in complex with USP7 at 2.9?? resolution. The interaction between the two proteins is primarily mediated by an acidic pocket in USP7 and Lysine residues within DNMT1’s KG linker. This intermolecular interaction is required for USP7-mediated stabilization of DNMT1. Acetylation of the KG linker Lysine residues impair DNMT1–USP7 interaction and promote the degradation of DNMT1. Treatment with HDAC inhibitors results in an increase in acetylated DNMT1 and decreased total DNMT1 protein. This negative correlation is observed in differentiated neuronal cells and pancreatic cancer cells. Our studies reveal that USP7-mediated stabilization of DNMT1 is regulated by acetylation and provide a structural basis for the design of inhibitors, targeting the DNMT1–USP7 interaction surface for therapeutic applications( Nature Communications, 2015). |
 |
| |
| Coordinated recognition of histone modifications by UHRF1 |
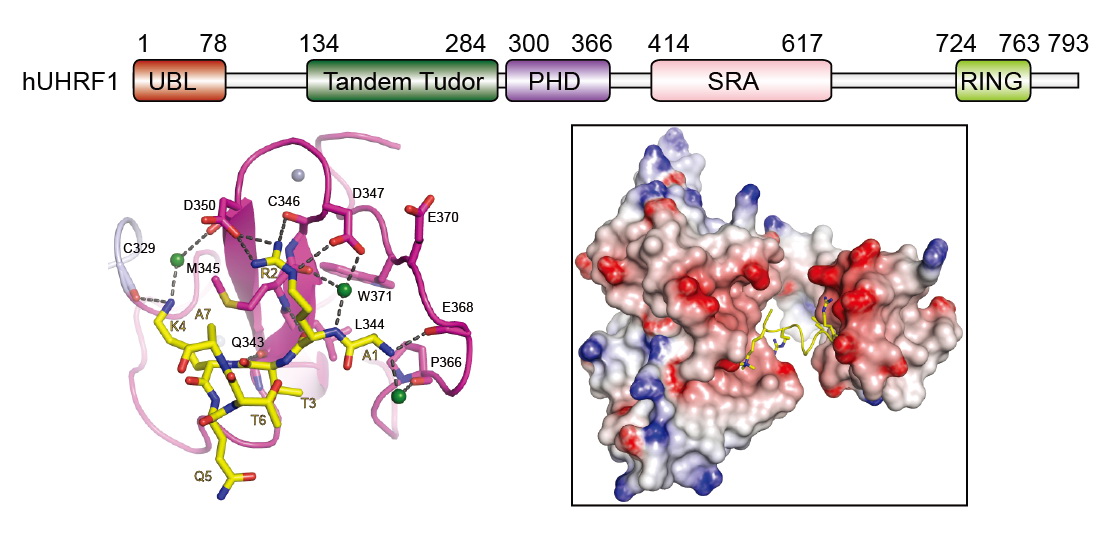 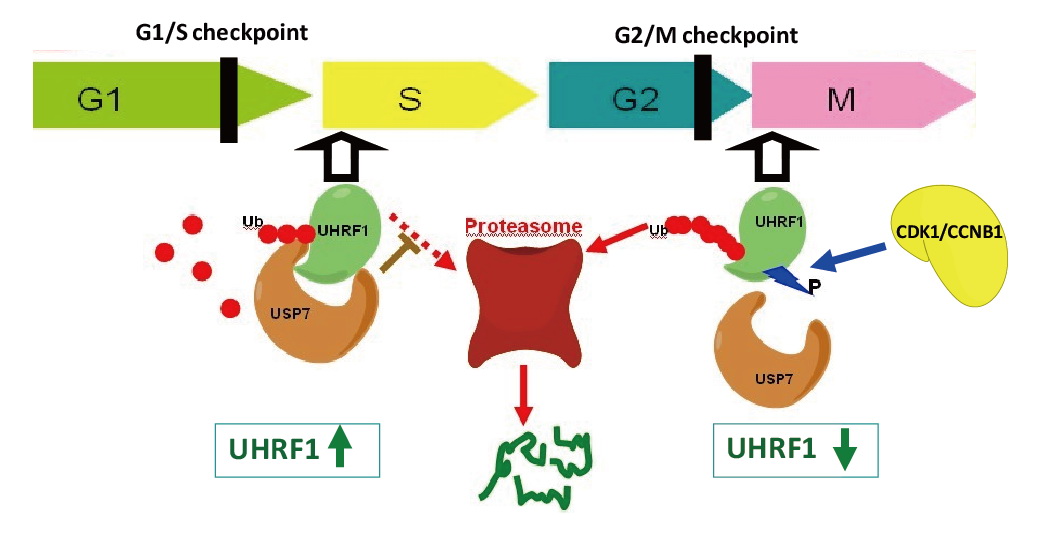 |
UHRF1 connects DNA and histone methylation. We demonstrate that PHD domain of UHRF1 recognizes unmodified histone H3, and H3R2 methylation disrupts the interaction and regulates UHRF1-mediated gene transcription ( Cell Res, 2011). We further show that TTD and PHD domains are essential for H3K9me3 recognition by UHRF1 ( JBC, 2013). Our studies further indicate that USP7 interacts with and stabilizes UHRF1, and phosphorylation of UHRF1 disrupts the interaction and leads to degradation of UHRF1 in M phase ( PNAS, 2012). |
 |
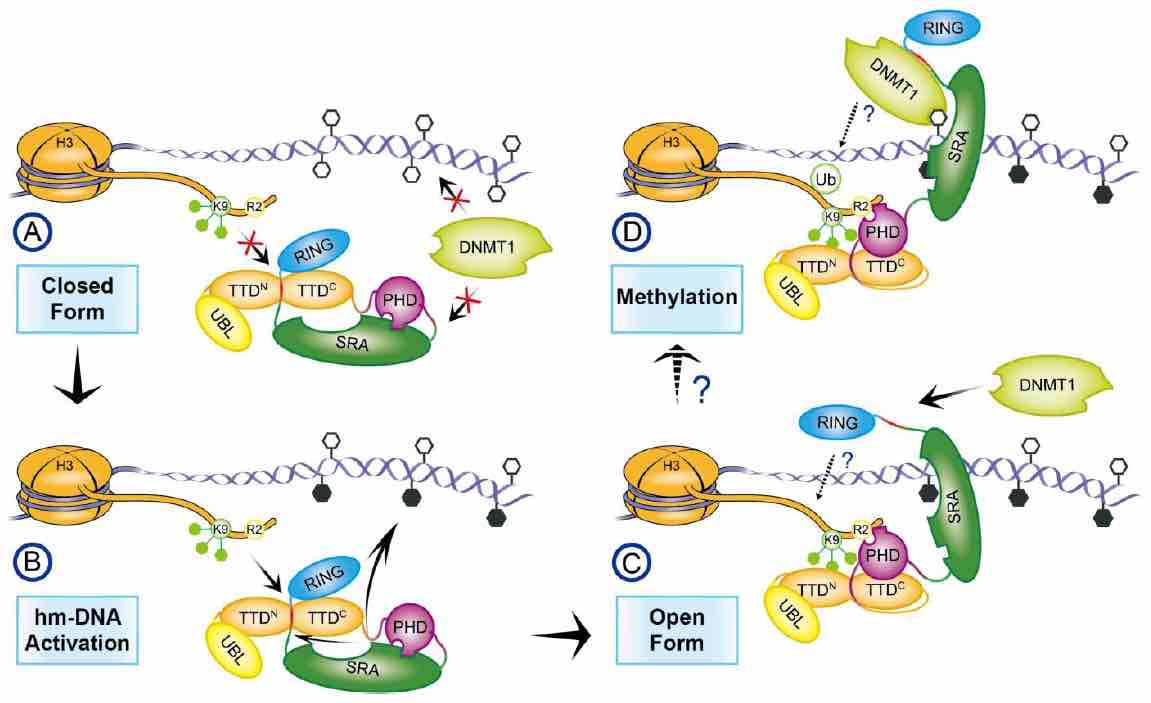 |
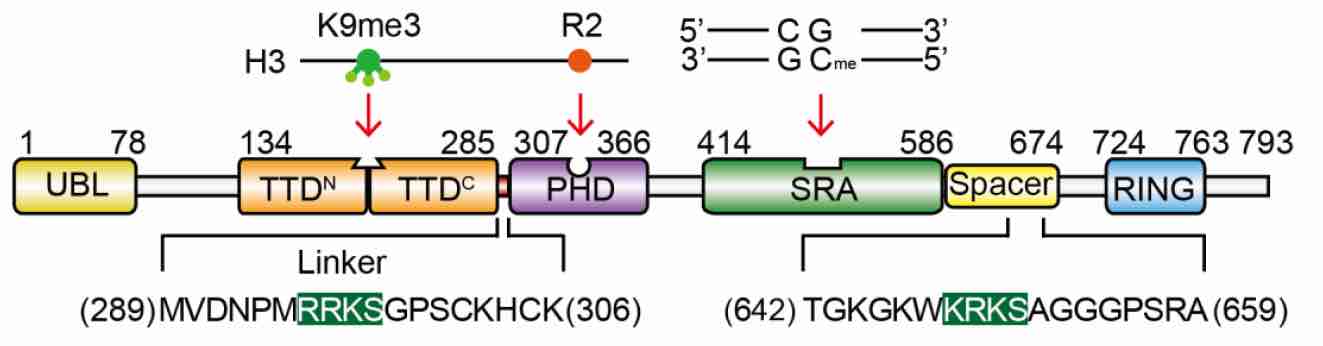
We found that UHRF1 adopts a closed conformation, in which a C-terminal region (Spacer) binds to the tandem Tudor domain (TTD) and inhibits H3K9me3 recognition, whereas the SET and RING associated (SRA) domain binds to the plant homeodomain (PHD) and inhibits H3R2 recognition. Hm-DNA impairs the intramolecular interactions and promotes H3K9me3 recognition by TTD-PHD. The Spacer also facilitates UHRF1-DNMT1 interaction and enhances hm-DNA-binding affinity of the SRA. When TTD-PHD binds to H3K9me3, SRA-Spacer may exist in a dynamic equilibrium: either recognizes hm-DNA or recruits DNMT1 to chromatin. Our study reveals the mechanism for regulation of H3K9me3 and hm-DNA recognition by URHF1 ( Nature Communications, 2016). |
Functional and Structural Studies of Histone Demethylases |
|
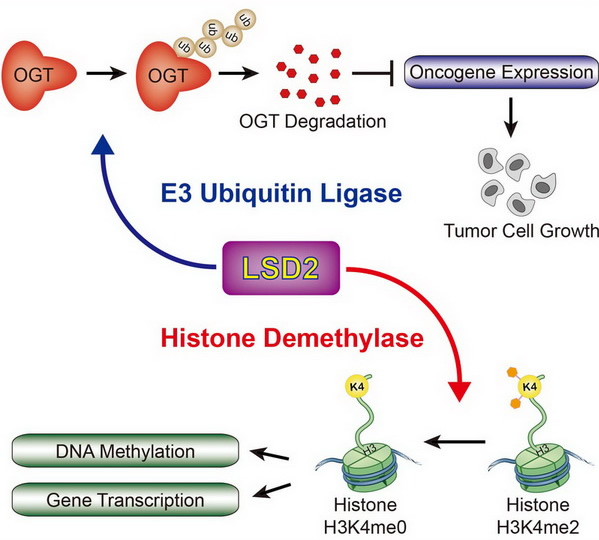 |
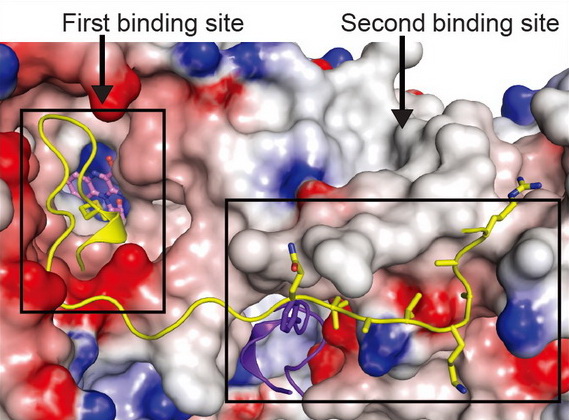
Structural and functional studies of LSD2, a H3K4me1/me2 demethylase, reveal that “second substrate binding site” is important for substrate recognition and enzymatic activity of LSD2 ( Cell Res, 2013). We also found that NPAC, an LSD2 binding protein, stimulates LSD2 enzymatic activity through enhancing the interaction between LSD2 and its histone substrate ( Mol Cell, 2013, Reviewed by Nature China). |
We found that LSD2 possesses an unexpected E3 ubiquitin ligase activity. LSD2 directly ubiquitylates and promotes degradation of OGT, and inhibits cancer cell growth in a manner dependent on its E3 ligase activity, but not demethylase activity. ( Mol Cell, 2015, Highlights in Science Signaling). |
|
|